Metylacja-specipc PCR (MSP) Kit
Cechy
■ Produkt ma zalety szybkości, prostoty, wysokiej czułości, silnej specyficzności i dobrej stabilności.
Specyfikacja
Rodzaj: Polimeraza DNA MSP
Szablon: <500 ng
Zastosowania: Nadaje się do specyficznej dla metylacji metody PCR (MSP) do analizy charakterystyki metylacji genomowego DNA
Wszystkie produkty można dostosować do potrzeb ODM/OEM. Dla szczegółów,kliknij opcję Dostosowana usługa (ODM/OEM)




1. Methylation-Specific PCR(MSP) Kit zastosowano do amplifikacji fragmentu 400 bp przy użyciu jako matrycy genomowego DNA traktowanego wodorosiarczynem z układem reakcyjnym 20 µl.

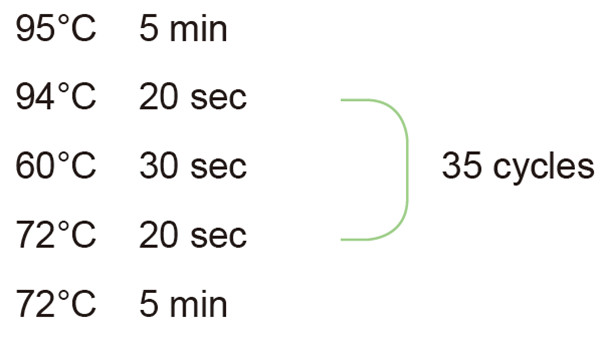
2. Ustawienie cyklu reakcji PCR
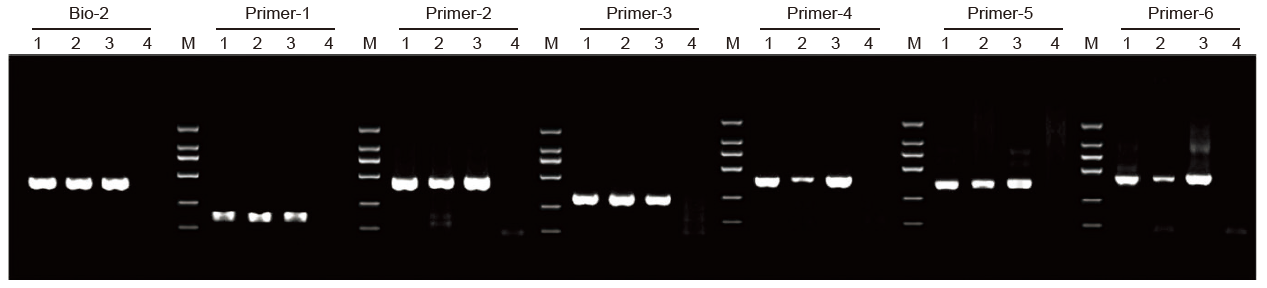 |
1: Próbki przetworzone przez Dostawcę A; 2: Próbki przetworzone przez Dostawcę B; 3: próbka traktowana TIANGEN 4: NTC; M: D2000 |
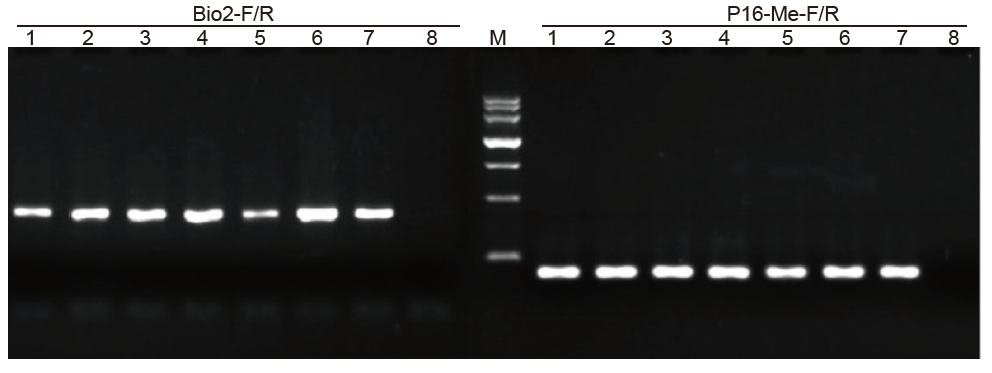 |
1-6: 6 powtórzeń; 7: Dostawca Przetworzona próbka; 8: NTC; Bio2-F/R i P16-Me-F/R: dwa startery detekcyjne. |
Szablon A-1
■ Matryca zawiera zanieczyszczenia białkowe lub inhibitory Taq itp. ——Oczyść matrycę DNA, usuń zanieczyszczenia białkowe lub ekstrahuj matryce DNA za pomocą zestawów do oczyszczania.
■ Denaturacja szablonu nie jest kompletna —— Odpowiednio zwiększyć temperaturę denaturacji i przedłużyć czas denaturacji.
■ Degradacja szablonu — — Ponownie przygotuj szablon.
Podkład A-2
■ Słaba jakość starterów —— Ponownie zsyntetyzuj starter.
■ Degradacja primera —— Rozdzielić primery o wysokim stężeniu do małej objętości w celu konserwacji. Unikaj wielokrotnego zamrażania i rozmrażania lub długotrwałego kriokonserwacji w temperaturze 4°C.
■ Niewłaściwe zaprojektowanie starterów (np. niewystarczająca długość startera, dimer utworzony między starterami itp.) - Przeprojektowanie starterów (unikaj tworzenia dimeru startera i struktury drugorzędowej)
A-3 Mg2+stężenie
■ Mg2+ stężenie jest zbyt niskie ——Właściwie zwiększyć Mg2+ stężenie: Optymalizacja Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
A-4 Temperatura wyżarzania
■ Wysoka temperatura annealingu wpływa na wiązanie startera i matrycy. —— Zmniejsz temperaturę wyżarzania i zoptymalizuj warunki z gradientem 2°C.
A-5 Czas przedłużenia
■ Krótki czas przedłużenia——Wydłużenie czasu przedłużenia.
Zjawiska: Próbki ujemne również wykazują prążki sekwencji docelowej.
A-1 Zanieczyszczenie PCR
■ Zanieczyszczenie krzyżowe sekwencji docelowej lub produktów amplifikacji ——Uważaj, aby nie odpipetować próbki zawierającej sekwencję docelową w próbce ujemnej ani nie wylać jej z probówki wirówkowej. Odczynniki lub sprzęt powinny być autoklawowane w celu wyeliminowania istniejących kwasów nukleinowych, a zanieczyszczenie powinno być stwierdzone poprzez eksperymenty z kontrolą ujemną.
■ Zanieczyszczenie odczynnikiem —— Podziel odczynniki i przechowuj w niskiej temperaturze.
A-2 Primer
■ Mg2+ stężenie jest zbyt niskie ——Właściwie zwiększyć Mg2+ stężenie: Optymalizacja Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
■ Niewłaściwy projekt startera i sekwencja docelowa ma homologię z sekwencją inną niż docelowa. —— Przeprojektuj podkłady.
Zjawiska: Prążki amplifikacji PCR są niezgodne z oczekiwaną wielkością, albo duże, albo małe, lub czasami występują zarówno specyficzne prążki amplifikacji, jak i nieswoiste prążki amplifikacji.
Podkład A-1
■ Słaba specyficzność startera
—— Przeprojektuj podkład.
■ Stężenie podkładu jest zbyt wysokie ——Właściwie zwiększ temperaturę denaturacji i przedłuż czas denaturacji.
A-2 Mg2+ stężenie
■ Mg2+ stężenie jest zbyt wysokie ——Właściwie zmniejsz stężenie Mg2+: Zoptymalizuj Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
A-3 Termostabilna polimeraza
■ Nadmierna ilość enzymu —— Odpowiednio zmniejsz ilość enzymu w odstępach 0,5 U.
A-4 Temperatura wyżarzania
■ Temperatura wyżarzania jest zbyt niska —— Odpowiednio zwiększyć temperaturę wyżarzania lub zastosować metodę wyżarzania dwuetapowego
A-5 cykli PCR
■ Za dużo cykli PCR —— Zmniejsz liczbę cykli PCR.
Podkład A-1——Słaba specyficzność ——Przeprojektuj starter, zmień położenie i długość startera, aby wzmocnić jego specyficzność; lub wykonaj zagnieżdżoną reakcję PCR.
A-2 Szablon DNA
—— Matryca nie jest czysta —— Oczyść matrycę lub wyodrębnij DNA za pomocą zestawów do oczyszczania.
A-3 Mg2+ stężenie
——Mg2+ stężenie jest zbyt wysokie ——Właściwie zmniejsz Mg2+ stężenie: Optymalizacja Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
A-4 dNTP
—— Stężenie dNTP jest zbyt wysokie —— Odpowiednio zmniejsz stężenie dNTP
A-5 Temperatura wyżarzania
——Zbyt niska temperatura wyżarzania ——Odpowiednio zwiększyć temperaturę wyżarzania
A-6 cykli
——Zbyt wiele cykli ——Zoptymalizuj liczbę cykli

Pierwszym krokiem jest wybór odpowiedniej polimerazy. Zwykła polimeraza Taq nie może dokonać korekty ze względu na brak aktywności 3'-5' egzonukleazy, a niedopasowanie znacznie zmniejszy wydajność wydłużania fragmentów. Dlatego zwykła polimeraza Taq nie może skutecznie amplifikować fragmentów docelowych większych niż 5 kb. Polimerazę Taq ze specjalną modyfikacją lub inną polimerazę o wysokiej wierności należy wybrać w celu poprawy wydajności wydłużania i zaspokojenia potrzeb amplifikacji długich fragmentów. Ponadto amplifikacja długich fragmentów wymaga również odpowiedniego dostosowania projektu startera, czasu denaturacji, czasu wydłużania, pH buforu itp. Zwykle startery o wielkości 18-24 pz mogą prowadzić do lepszej wydajności. Aby zapobiec uszkodzeniu matrycy, czas denaturacji w 94°C należy skrócić do 30 sekund lub mniej na cykl, a czas wzrostu temperatury do 94°C przed amplifikacją powinien być krótszy niż 1 min. Ponadto ustawienie temperatury wydłużania na około 68°C i zaprojektowanie czasu wydłużania na poziomie 1 kb/min może zapewnić skuteczną amplifikację długich fragmentów.
Wskaźnik błędu amplifikacji PCR można zmniejszyć, stosując różne polimerazy DNA o wysokiej wierności. Spośród wszystkich dotychczas znalezionych polimeraz DNA Taq, enzym Pfu ma najniższy wskaźnik błędów i najwyższą wierność (patrz załączona tabela). Oprócz selekcji enzymów, naukowcy mogą jeszcze bardziej zmniejszyć szybkość mutacji PCR poprzez optymalizację warunków reakcji, w tym optymalizację składu buforu, stężenia termostabilnej polimerazy i optymalizację liczby cykli PCR.
Kategorie produktów
DLACZEGO WŁAŚNIE MY
Od momentu powstania nasza fabryka opracowuje produkty pierwszej światowej klasy, przestrzegając zasady
najpierw jakość. Nasze produkty zyskały doskonałą renomę w branży i cenne zaufanie wśród nowych i starych klientów.
- Tel: +86 010-59822688
- Budynek 5, nr 86, Shuangying West Road, dzielnica Changping, Pekin.
- ludzie@tiangen.com



