2×Taq Platinum PCR Mix
Definicja aktywności
1 jednostka (U) Aktywność polimerazy Taq Platinum DNA jest zdefiniowana jako ilość enzymu wymagana do włączenia 10 nmol deoksynukleotydów do substancji nierozpuszczalnych w kwasach w temperaturze 74°C w ciągu 30 minut przy użyciu aktywowanego DNA spermy łososia jako matrycy/startera.
Kontrola jakości
Czystość przez detekcję SDS-PAGE wynosi ponad 99%; Nie wykryto aktywności egzogennej nukleazy; Pojedyncza kopia genu w ludzkim genomie może być skutecznie amplifikowana; Brak znaczącej zmiany aktywności przy przechowywaniu w temperaturze pokojowej przez tydzień.
Główne parametry techniczne
Posiada aktywność egzonukleazy 5′-3′ i aktywność egzonukleazy 3′-5′, a jej wierność jest obok polimerazy Pfu. Szybkość wydłużania Taq Platinum Polymerase jest większa niż polimerazy Pfu, a wydajność amplifikacji jest wyższa. Produkty PCR można bezpośrednio ligować z tępym końcem lub klonować za pomocą wektora TA. Jeśli wydajność klonowania wymaga poprawy, zaleca się najpierw oczyszczenie i dodanie nawisów 3'-dA przed klonowaniem do wektora TA.
Jedna tubka Taq Platinum MasterMix (krajowa certyfikacja produktów high-tech)
■ Taq Platinum MasterMix ma poprawioną specyficzność i czułość reakcji PCR i może amplifikować złożone matryce o wysokiej zawartości GC, strukturze drugorzędowej i tym podobnych. Można amplifikować zaledwie 2 kopie docelowego szablonu, zapewniając dokładniejsze wyniki eksperymentalne.
■ Unikalna formuła Taq Platinum MasterMix sprawia, że cały układ reakcyjny jest bardzo stabilny, a na jego aktywność nie wpłynie wielokrotne zamrażanie-rozmrażanie lub długotrwałe przechowywanie w temperaturze 4°C.
■ Stabilny i wydajny, wstępnie przygotowany mieszany roztwór PCR może sprawić, że operacja będzie szybka i prosta, znacznie zmniejszając pracochłonność i błąd próbkowania. Mieszanka zawiera również wysokowydajny wzmacniacz i optymalizator PCR, co zmniejsza wymagania dotyczące warunków PCR.
■ Ten produkt zawiera systemy zawierające barwniki i bez barwników. Produkty MasterMix zawierające barwnik mogą być poddane bezpośredniej elektroforezie po reakcji PCR, bez dodawania buforu ładującego.
Aplikacje
Może zastąpić polimerazę Pfu w celu amplifikacji produktów o wysokiej wierności ze złożonych matryc, takich jak genomy, i nadaje się do zastosowań takich jak klonowanie genów ekspresyjnych, mutacje specyficzne dla miejsca i analiza polimorfizmu pojedynczego nukleotydu (SNP) itp.
Środki ostrożności przy projektowaniu starterów PCR:
Długość podkładu wynosi zwykle 20-25 mer. Jednak przy wykonywaniu PCR na długich fragmentach długość startera powinna być zwiększona do 30-35 merów.
■ Nie ma komplementarnego parowania między dwoma starterami, szczególnie dla ostatnich 3 zasad na końcu 3′.
■ Zawartość GC powinna wynosić 50-60% i unikać lokalnego bogatego GC lub AT. Aby starter i matryca wiązały się stabilnie, unikaj struktury bogatej w AT na końcu 3'.
■ Unikaj podkładu, aby utworzyć strukturę drugorzędową.
■ Wybierz dwa podkłady o temperaturach Tm zbliżonych do siebie.
Obliczanie wartości Tm starterów do PCR:
■ Gdy podkład ma mniej niż 20 merów: Tm=2°C×(A+T)+4°C×(G+C).
■ Gdy starter ma więcej niż 20 merów: Tm=81,5+0,41×(GC%)-600/L, gdzie L jest długością startera.
■ Ustawić temperaturę wyżarzania na (Tm-5)°C.
Wejście startera PCR
Odpowiednie końcowe stężenie starterów można wybrać między 0,1 μM a 1,0 μM. Zbyt niskie stężenie startera prowadzi do niskiej wydajności produktów amplifikacji, podczas gdy zbyt wysokie stężenie startera jest bardziej podatne na nieswoistą amplifikację. Zwykle, gdy ilość matrycowego DNA jest duża lub gdy jako matryca stosuje się złożony matrycowy DNA (taki jak DNA genomu ludzkiego), stężenie startera powinno być niższe. Gdy ilość matrycowego DNA jest mała lub gdy jako matryca stosuje się prosty matrycowy DNA (np. plazmidowy DNA, itp.), stężenie startera powinno być wyższe.
Wszystkie produkty można dostosować do potrzeb ODM/OEM. Dla szczegółów,kliknij opcję Dostosowana usługa (ODM/OEM)




 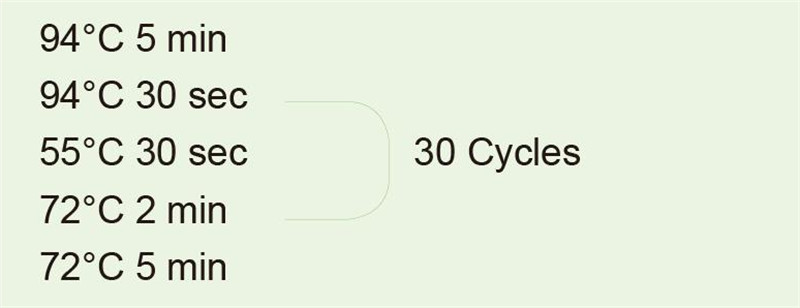 |
Użyj genomowego DNA jako matrycy do amplifikacji fragmentu 1 kb. Po reakcji PCR weź 5 μl do detekcji elektroforetycznej. |
Szablon A-1
■ Matryca zawiera zanieczyszczenia białkowe lub inhibitory Taq itp. ——Oczyść matrycę DNA, usuń zanieczyszczenia białkowe lub ekstrahuj matryce DNA za pomocą zestawów do oczyszczania.
■ Denaturacja szablonu nie jest kompletna —— Odpowiednio zwiększyć temperaturę denaturacji i przedłużyć czas denaturacji.
■ Degradacja szablonu — — Ponownie przygotuj szablon.
Podkład A-2
■ Słaba jakość starterów —— Ponownie zsyntetyzuj starter.
■ Degradacja primera —— Rozdzielić primery o wysokim stężeniu do małej objętości w celu konserwacji. Unikaj wielokrotnego zamrażania i rozmrażania lub długotrwałego kriokonserwacji w temperaturze 4°C.
■ Niewłaściwe zaprojektowanie starterów (np. niewystarczająca długość startera, dimer utworzony między starterami itp.) - Przeprojektowanie starterów (unikaj tworzenia dimeru startera i struktury drugorzędowej)
A-3 Mg2+stężenie
■ Mg2+ stężenie jest zbyt niskie ——Właściwie zwiększyć Mg2+ stężenie: Optymalizacja Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
A-4 Temperatura wyżarzania
■ Wysoka temperatura annealingu wpływa na wiązanie startera i matrycy. —— Zmniejsz temperaturę wyżarzania i zoptymalizuj warunki z gradientem 2°C.
A-5 Czas przedłużenia
■ Krótki czas przedłużenia——Wydłużenie czasu przedłużenia.
Zjawiska: Próbki ujemne również wykazują prążki sekwencji docelowej.
A-1 Zanieczyszczenie PCR
■ Zanieczyszczenie krzyżowe sekwencji docelowej lub produktów amplifikacji ——Uważaj, aby nie odpipetować próbki zawierającej sekwencję docelową w próbce ujemnej ani nie wylać jej z probówki wirówkowej. Odczynniki lub sprzęt powinny być autoklawowane w celu wyeliminowania istniejących kwasów nukleinowych, a zanieczyszczenie powinno być stwierdzone poprzez eksperymenty z kontrolą ujemną.
■ Zanieczyszczenie odczynnikiem —— Podziel odczynniki i przechowuj w niskiej temperaturze.
A-2 Primer
■ Mg2+ stężenie jest zbyt niskie ——Właściwie zwiększyć Mg2+ stężenie: Optymalizacja Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
■ Niewłaściwy projekt startera i sekwencja docelowa ma homologię z sekwencją inną niż docelowa. —— Przeprojektuj podkłady.
Zjawiska: Prążki amplifikacji PCR są niezgodne z oczekiwaną wielkością, albo duże, albo małe, lub czasami występują zarówno specyficzne prążki amplifikacji, jak i nieswoiste prążki amplifikacji.
Podkład A-1
■ Słaba specyficzność startera
—— Przeprojektuj podkład.
■ Stężenie podkładu jest zbyt wysokie ——Właściwie zwiększ temperaturę denaturacji i przedłuż czas denaturacji.
A-2 Mg2+ stężenie
■ Mg2+ stężenie jest zbyt wysokie ——Właściwie zmniejsz stężenie Mg2+: Zoptymalizuj Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
A-3 Termostabilna polimeraza
■ Nadmierna ilość enzymu —— Odpowiednio zmniejsz ilość enzymu w odstępach 0,5 U.
A-4 Temperatura wyżarzania
■ Temperatura wyżarzania jest zbyt niska —— Odpowiednio zwiększyć temperaturę wyżarzania lub zastosować metodę wyżarzania dwuetapowego
A-5 cykli PCR
■ Za dużo cykli PCR —— Zmniejsz liczbę cykli PCR.
Podkład A-1——Słaba specyficzność ——Przeprojektuj starter, zmień położenie i długość startera, aby wzmocnić jego specyficzność; lub wykonaj zagnieżdżoną reakcję PCR.
A-2 Szablon DNA
—— Matryca nie jest czysta —— Oczyść matrycę lub wyodrębnij DNA za pomocą zestawów do oczyszczania.
A-3 Mg2+ stężenie
——Mg2+ stężenie jest zbyt wysokie ——Właściwie zmniejsz Mg2+ stężenie: Optymalizacja Mg2+ stężenie poprzez serię reakcji od 1 mM do 3 mM w odstępie 0,5 mM w celu określenia optymalnego Mg2+ stężenie dla każdej matrycy i startera.
A-4 dNTP
—— Stężenie dNTP jest zbyt wysokie —— Odpowiednio zmniejsz stężenie dNTP
A-5 Temperatura wyżarzania
——Zbyt niska temperatura wyżarzania ——Odpowiednio zwiększyć temperaturę wyżarzania
A-6 cykli
——Zbyt wiele cykli ——Zoptymalizuj liczbę cykli

Pierwszym krokiem jest wybór odpowiedniej polimerazy. Zwykła polimeraza Taq nie może dokonać korekty ze względu na brak aktywności 3'-5' egzonukleazy, a niedopasowanie znacznie zmniejszy wydajność wydłużania fragmentów. Dlatego zwykła polimeraza Taq nie może skutecznie amplifikować fragmentów docelowych większych niż 5 kb. Polimerazę Taq ze specjalną modyfikacją lub inną polimerazę o wysokiej wierności należy wybrać w celu poprawy wydajności wydłużania i zaspokojenia potrzeb amplifikacji długich fragmentów. Ponadto amplifikacja długich fragmentów wymaga również odpowiedniego dostosowania projektu startera, czasu denaturacji, czasu wydłużania, pH buforu itp. Zwykle startery o wielkości 18-24 pz mogą prowadzić do lepszej wydajności. Aby zapobiec uszkodzeniu matrycy, czas denaturacji w 94°C należy skrócić do 30 sekund lub mniej na cykl, a czas wzrostu temperatury do 94°C przed amplifikacją powinien być krótszy niż 1 min. Ponadto ustawienie temperatury wydłużania na około 68°C i zaprojektowanie czasu wydłużania na poziomie 1 kb/min może zapewnić skuteczną amplifikację długich fragmentów.
Wskaźnik błędu amplifikacji PCR można zmniejszyć, stosując różne polimerazy DNA o wysokiej wierności. Spośród wszystkich dotychczas znalezionych polimeraz DNA Taq, enzym Pfu ma najniższy wskaźnik błędów i najwyższą wierność (patrz załączona tabela). Oprócz selekcji enzymów, naukowcy mogą jeszcze bardziej zmniejszyć szybkość mutacji PCR poprzez optymalizację warunków reakcji, w tym optymalizację składu buforu, stężenia termostabilnej polimerazy i optymalizację liczby cykli PCR.
Kategorie produktów
DLACZEGO WŁAŚNIE MY
Od momentu powstania nasza fabryka opracowuje produkty pierwszej światowej klasy, przestrzegając zasady
najpierw jakość. Nasze produkty zyskały doskonałą renomę w branży i cenne zaufanie wśród nowych i starych klientów.
- Tel: +86 010-59822688
- Budynek 5, nr 86, Shuangying West Road, dzielnica Changping, Pekin.
- ludzie@tiangen.com



