Zestaw do oznaczania ilościowego DNA TIANSeq (Illumina)
Cechy
■ Prosty i szybki: zestaw zawiera wszystkie odczynniki do oznaczania ilościowego biblioteki. Jest prosty i szybki w obsłudze oraz ma dobrą kompatybilność z zestawami konstrukcyjnymi biblioteki nadrzędnej.
■ Dokładna ocena ilościowa: Odczynnik wykrywający ma wysoką wydajność amplifikacji i dobrą specyficzność oraz ma dobrą zależność liniową w zakresie 0,0002 pM 20 pM.
■ Szerokie zastosowanie: odczynnik qPCR dostarczany przez zestaw ma dobrą uniwersalność dla fragmentów o różnej zawartości i wielkości GC.
Aplikacje
Określ stężenie wysokoprzepustowej biblioteki do sekwencjonowania przed sekwencjonowaniem na platformie Illumina.
Wszystkie produkty można dostosować do potrzeb ODM/OEM. Dla szczegółów,kliknij opcję Dostosowana usługa (ODM/OEM)




Obecnie technologia sekwencjonowania o wysokiej przepustowości opiera się głównie na technologii sekwencjonowania nowej generacji. Ponieważ długość odczytu technologii sekwencjonowania nowej generacji jest ograniczona, musimy rozbić sekwencję pełnej długości na biblioteki małych fragmentów, aby zsekwencjonować. W zależności od potrzeb różnych eksperymentów sekwencjonowania, zwykle wybieramy sekwencjonowanie single-ended lub double-ended sekwencjonowanie. Obecnie fragmenty DNA biblioteki sekwencjonowania nowej generacji są na ogół rozmieszczone w zakresie 200-800 pz.
a) DNA jest niskiej jakości i zawiera inhibitory. Używaj wysokiej jakości próbek DNA, aby uniknąć hamowania aktywności enzymów.
b) Ilość próbki DNA jest niewystarczająca przy użyciu metody wolnej od PCR do konstruowania biblioteki DNA. Gdy ilość pofragmentowanego DNA przekracza 50 ng, podczas procesu konstrukcji biblioteki można selektywnie przeprowadzić przepływ pracy bez PCR. Jeśli liczba kopii biblioteki jest zbyt niska, aby można ją było bezpośrednio zsekwencjonować, bibliotekę DNA można amplifikować metodą PCR po ligacji adaptera.
c) Zanieczyszczenie RNA prowadzi do niedokładnej początkowej oceny ilościowej DNA Zanieczyszczenie RNA może występować w procesie oczyszczania genomowego DNA, co może prowadzić do niedokładnej oceny ilościowej DNA i niewystarczającego obciążenia DNA podczas konstrukcji biblioteki. RNA można usunąć przez traktowanie RNazą.
A-1
a) Pojawiają się małe fragmenty (60 bp-120 bp) Małe fragmenty są zazwyczaj fragmentami adaptorów lub dimerami utworzonymi przez adaptory. Oczyszczanie za pomocą kulek magnetycznych Agencourt AMPure XP może skutecznie usunąć te fragmenty adaptera i zapewnić jakość sekwencjonowania.
b) Duże fragmenty pojawiają się w bibliotece po amplifikacji PCR Wielkość fragmentu DNA z biblioteki zwiększy się o 120 bp po ligacji adaptera. Jeśli po ligacji adaptera fragment DNA zwiększa się o więcej niż 120 pz, może to być spowodowane nieprawidłową amplifikacją fragmentu lub nadmierną amplifikacją PCR. Zmniejszenie liczby cykli PCR może zapobiec tej sytuacji.
c) Nieprawidłowa wielkość fragmentów DNA biblioteki po ligacji adaptera Długość adaptera w tym zestawie wynosi 60 pz. Gdy dwa końce fragmentu zostaną zligowane z adapterami, długość wzrośnie tylko o 120 pz. W przypadku korzystania z adaptera innego niż dostarczony w tym zestawie należy skontaktować się z dostawcą w celu podania odpowiednich informacji, takich jak długość adaptera. Upewnij się, że przepływ pracy i działanie eksperymentu są zgodne z krokami opisanymi w instrukcji.
d) Nieprawidłowa wielkość fragmentu DNA przed ligacją adaptera Przyczyną tego problemu mogą być złe warunki reakcji podczas fragmentacji DNA. Dla różnych danych wejściowych DNA należy stosować różne czasy reakcji. Jeśli wkład DNA jest większy niż 10 ng, jako czas rozpoczęcia optymalizacji zalecamy wybrać czas reakcji 12 min, a wielkość fragmentów wytworzonych w tym czasie mieści się głównie w zakresie 300-500 pz. Użytkownicy mogą zwiększać lub zmniejszać długość fragmentów DNA o 2-4 min zgodnie z własnymi wymaganiami, aby zoptymalizować fragmenty DNA o wymaganym rozmiarze.
A-2
a) Czas fragmentacji nie jest zoptymalizowany Jeśli pofragmentowane DNA jest za małe lub za duże, należy zapoznać się z Wytycznymi doboru czasu fragmentacji zawartymi w instrukcji, aby określić czas reakcji i wykorzystać ten punkt czasowy jako kontrolę, dodatkowo ustawić system reakcyjny do przedłużenia lub skrócenia 3 min w celu dokładniejszej regulacji czasu rozdrabniania.
A-3
Nieprawidłowy rozkład wielkości DNA po fragmentacji
a) Nieprawidłowa metoda rozmrażania odczynnika fragmentującego lub odczynnik nie jest całkowicie wymieszany po rozmrożeniu. Rozmrozić odczynnik 5× Fragmentation Enzyme Mix na lodzie. Po rozmrożeniu, wymieszaj równomiernie odczynnik, delikatnie stukając dnem probówki. Nie worteksować odczynnika!
b) Próbka wejściowa DNA zawiera EDTA lub inne zanieczyszczenia Zubożenie jonów soli i czynników chelatujących na etapie oczyszczania DNA jest szczególnie ważne dla powodzenia eksperymentu. Jeśli DNA jest rozpuszczone w 1×TE, użyj metody podanej w instrukcji, aby przeprowadzić fragmentację. Jeśli stężenie EDTA w roztworze jest niepewne, zaleca się oczyszczenie DNA i rozpuszczenie go w wodzie dejonizowanej do dalszej reakcji.
c) Niedokładna początkowa ocena ilościowa DNA Wielkość pofragmentowanego DNA jest ściśle związana z ilością wprowadzonego DNA. Przed obróbką fragmentacji, dokładna ocena ilościowa DNA przy użyciu Qubit, Picogreen i innych metod jest niezbędna do określenia dokładnej ilości DNA w układzie reakcyjnym.
d) Przygotowanie układu reakcyjnego nie jest zgodne z instrukcją Przygotowanie rozdrobnionego układu reakcyjnego musi być przeprowadzone na lodzie ściśle według instrukcji. Aby zapewnić jak najlepszy efekt, wszystkie składniki reakcji należy umieścić na lodzie, a przygotowanie układu reakcyjnego przeprowadzić po całkowitym schłodzeniu. Po zakończeniu przygotowania należy przetrzepać lub pipetować, aby dokładnie wymieszać. Nie wiruj!
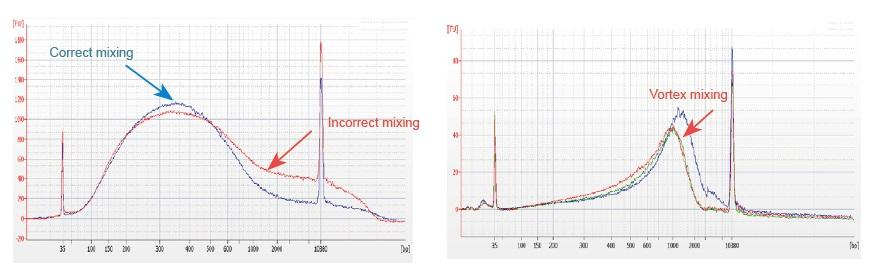
1. Niewłaściwa metoda mieszania (wir, gwałtowne oscylacje itp.) spowoduje nieprawidłową dystrybucję fragmentów biblioteki (jak pokazano na poniższym rysunku), wpływając tym samym na jakość biblioteki. Dlatego przygotowując roztwór reakcyjny Fragmentation Mix, należy delikatnie pipetować w górę iw dół w celu wymieszania lub użyć opuszki palca, aby równomiernie przetrzepać i wymieszać. Uważaj, aby nie mieszać z wirami.
2. Do budowy biblioteki należy użyć DNA o wysokiej czystości
■ Dobra integralność DNA: pasmo elektroforetyczne ma ponad 30 kb, bez ogonków
■ OD260/230: >1,5
■ OD260/280: 1,7-1,9
3. Ilość wejściowa DNA musi być dokładna Do ilościowego oznaczania DNA sugeruje się stosowanie metod Qubit i PicoGreen, a nie Nanodrop.
4. Należy oznaczyć zawartość EDTA w roztworze DNA EDTA ma duży wpływ na reakcję fragmentacji. Jeśli zawartość EDTA jest wysoka, oczyszczanie DNA należy przeprowadzić przed kolejnym testem.
5. Roztwór reakcji fragmentacji należy przygotować na lodzie Proces fragmentacji jest wrażliwy na temperaturę i czas reakcji (zwłaszcza po dodaniu wzmacniacza). W celu zapewnienia dokładności czasu reakcji należy przygotować układ reakcyjny na lodzie.
6. Czas reakcji fragmentacji musi być dokładny Czas reakcji etapu fragmentacji bezpośrednio wpłynie na wielkość produktów fragmentów, wpływając w ten sposób na rozkład wielkości fragmentów DNA w bibliotece.
1. Jaki rodzaj próbki ma zastosowanie do tego zestawu?
Odpowiednim typem próbki tego zestawu może być całkowity RNA lub oczyszczony mRNA o dobrej integralności RNA. Jeśli do skonstruowania biblioteki używany jest całkowity RNA, zaleca się najpierw użycie zestawu do usuwania rRNA (nr kat. 4992363/4992364/4992391) w celu usunięcia rRNA.
2. Czy próbki FFPE mogą być użyte do budowy biblioteki za pomocą tego zestawu?
mRNA w próbkach FFPE ulegnie degradacji do pewnego stopnia ze względną słabą integralnością. Przy stosowaniu tego zestawu do budowy biblioteki zaleca się optymalizację czasu fragmentacji (skrócenie czasu fragmentacji lub niewykonanie fragmentacji).
3. Korzystając z kroku wyboru rozmiaru podanego w instrukcji produktu, co może spowodować, że wstawiony segment będzie wyglądał na niewielkie odchylenie?
Wybór rozmiaru powinien odbywać się ściśle zgodnie z etapem wyboru rozmiaru opisanym w niniejszej instrukcji produktu. Jeśli występuje odchylenie, przyczyną może być to, że kulki magnetyczne nie są wyważone w temperaturze pokojowej lub nie są w pełni wymieszane, pipeta nie jest dokładna lub płyn pozostaje w końcówce. Do eksperymentu zaleca się stosowanie końcówek o niskiej adsorpcji.
4. Dobór adapterów w budowie bibliotek
Zestaw do konstrukcji biblioteki nie zawiera odczynnika adaptera i zaleca się używanie tego zestawu razem z adapterem TIANSeq Single-Index (Illumina) (4992641/4992642/4992378).
5. Kontrola jakości biblioteki
Wykrywanie ilościowe biblioteki: Qubit i qPCR są używane do określenia odpowiednio stężenia masowego i stężenia molowego biblioteki. Operacja jest ściśle zgodna z instrukcją produktu. Stężenie biblioteki będzie ogólnie spełniało wymagania sekwencjonowania NGS. Wykrywanie zakresu dystrybucji biblioteki: Stosowanie Agilent 2100 Bioanalyzer do wykrywania zakresu dystrybucji biblioteki.
6. Wybór numeru cyklu amplifikacji
Zgodnie z instrukcją liczba cykli PCR wynosi 6-12, a liczba potrzebnych cykli PCR powinna być dobrana zgodnie z wkładem próbki. W bibliotekach wysokowydajnych nadamplifikacja zwykle występuje w różnym stopniu, co objawia się nieco większym pikiem po piku zakresu docelowego w detekcji Agilent 2100 Bioanalyzer lub wykryte stężenie Qubit jest niższe niż qPCR. Łagodna amplifikacja jest zjawiskiem normalnym, które nie wpływa na sekwencjonowanie biblioteki i późniejszą analizę danych.
7. W profilu wykrywania Agilent 2100 Bioanalyzer pojawiają się kolce
Pojawienie się skoków w detekcji Agilent 2100 Bioanalyzer jest spowodowane nierównomierną fragmentacją próbek, gdzie będzie więcej fragmentów o określonej wielkości, co stanie się bardziej widoczne po wzbogaceniu metodą PCR. W takim przypadku sugeruje się, aby nie dokonywać selekcji wielkości, tj. ustawić warunki fragmentacji na 94°C przez 15 min inkubacji, gdzie rozkład fragmentów jest mały i skoncentrowany, a jednorodność można poprawić.
Kategorie produktów
DLACZEGO WŁAŚNIE MY
Od momentu powstania nasza fabryka opracowuje produkty pierwszej światowej klasy, przestrzegając zasady
najpierw jakość. Nasze produkty zyskały doskonałą renomę w branży i cenne zaufanie wśród nowych i starych klientów.
- Tel: +86 010-59822688
- Budynek 5, nr 86, Shuangying West Road, dzielnica Changping, Pekin.
- ludzie@tiangen.com



